组装步骤和分枝方法¶
一个典型的工作流程,从输入文件-fastq格式数据到输出文件-assembled输出文件主要有七个步骤。将这些步骤独立的分开来的主要是为了增强断点重续功能;另外,还能够在不同支点进行灵活分支-用不同参数进行计算并最终将他们进行整合。
基本流程¶
使用ipyrad最简便的方法,即运行设定好的参数脚本文件。第一步将不同的片段reads分类到各个样本中,第2-5步处理每个样本内部的数据,第6步在不同样本间聚类,第7步过滤所得数据及形成下游分析所需格式。
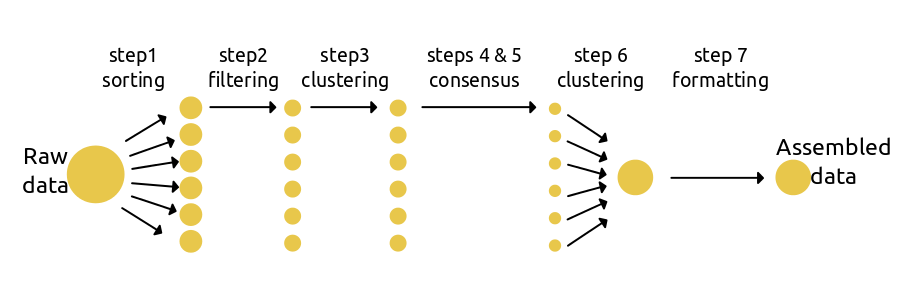
基本流程的运行代码很简单:
## 产生一个原始的参数文件
>>> ipyrad -n data1
## 用读者自己的文件编辑器对参数文件进行编辑
## ... 编辑ing params-data1.txt
## 用该参数文件运行步骤1-7
>>> ipyrad -p params-data1.txt -s 1234567
分支工作流程¶
使用ipyrad更高效的方法是将多数据集进行分支,用不同的参数文件进行计算。下面的原理图展示了在第三步进行分支计算的例子。新的分支将会延续之前的文件路径,但是能够继续用新的参数文件进行计算。分支计算在当前文件夹中并不会产生许多拷贝,所以不会占用很多的时间和空间。强烈建议在需要进行新的参数计算的时候使用这种方法。
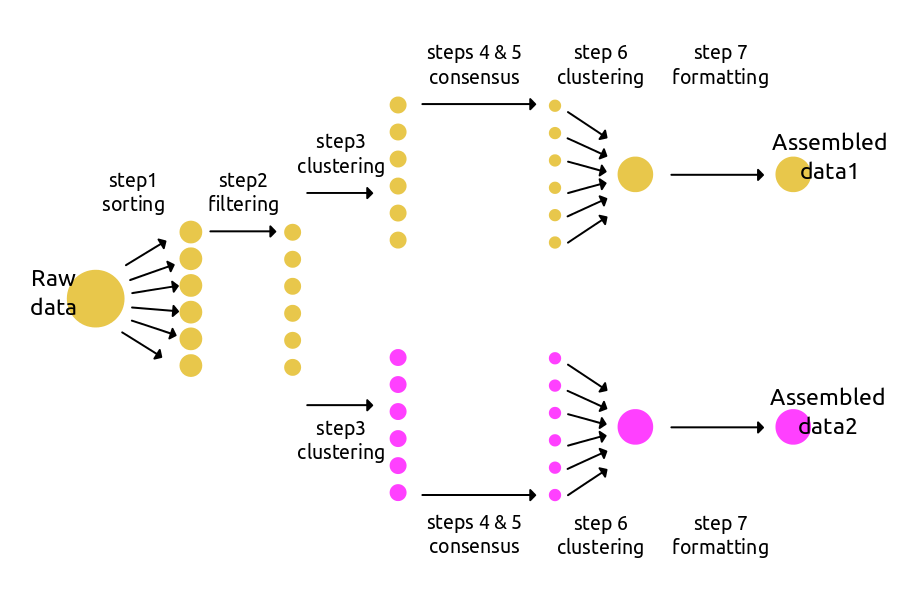
运行分支工作流程的代码只是比基础工作流程复杂了一点点。可在高级指导(Advanced tutorial-CLI) 和操作指南模块中找到更多的分支例子。
## 产生一个原始的组装过程和参数文件,这里将其命名为'data1'
>>> ipyrad -n data1
## 编辑data1的参数文件
## ... 编辑ing params-data1.txt
## 用该参数文件运行步骤1-2
>>> ipyrad -p params-data1.txt -s 12
## 在运行第三步之前先产生一个新的分支,命名为'data2'
>>> ipyrad -p params-data1.txt -b data2
## 编辑data2的参数文件
## ... 编辑ing params-data2.txt
## 将以上两者都运行步骤3-7
>>> ipyrad -p params-data1.txt -s 34567
>>> ipyrad -p params-data2.txt -s 34567
通过分支增加或减少样本¶
分支另一个用法在于从一个组装过程中,增加或减少样本量,或者分析所有样本中的某个子集,或去除部分地覆盖度的样本。通过ipyrad中 branching 和 merging 功能可以很容易地实现。为了减少样本而调用分支过程会将原来的组装过程保存为备份,这使得读者可以尝试很多不同的分支而不影响原来的计算。
用ipyrad CLI的一个例子:
## 分支,仅保留data1组装的3个样本
>>> ipyrad -n data1 -b data2 1A0 1B0 1C0
## 和/或,分支,仅除去data1组装的三个样本
>>> ipyrad -n data1 -b data3 - 1A0 1B0 1C0
使用ipyrad Python API的例子:
## 分支,仅保留data1组装的3个样本
>>> data1.branch("data2", subsamples=["1A0", "1B0", "1C0"])
## 和/或,分支,仅除去data1组装的三个样本
>>> keep_list = [i for i in data1.samples.keys() if i not in ["1A0", "1B0", "1C0"]]
>>> data1.branch("data3", subsamples=keep_list)
七个步骤¶
1.解编/加载fastq文件¶
步骤1主要是加载序列文件至已命名的Assembly并将reads分配到样本(个体)中。若数据未进行解编,第一步将使用barcodes文件进行解编,否则,就进行简单的数据归类。
以下是step 1中 可能用到 的和必须(带*的)参数:
2.过滤/编辑reads¶
根据碱基质量过滤片段,标记一些列低于限定值的碱基为N,N的数量高于设定值则将该reads抛弃。该阈值在 phred_Qscore_offset中设置。一个可选择的过滤器可以用来移除adapters/primers(见参数文件中的filter_adapters),也有一个可供选择的过滤器来清除边缘低质量的序列(见参数文件中的 edit_cutsites)
以下是step 2中 可能用到的 和必须(带*的)参数:
3.个体内聚类¶
首先将步骤2中的序列进行再复制,记录观察到的 unique read 次数。若数据为双末端,则用vserch合并配对重叠的reads。结果数据进行从头组装(用vsearch)或map到参考序列上(用smalt & bedtools),这取决于选择的组装方法。这二者都用muscle根据序列相似性聚类获得。
以下是step 3中 可能用到的 和必须(带*的)参数:
4. 联合估计杂合性和错误率¶
根据聚类的reads计算位点模式,据其估计测序错误率及杂合性。此估计用于step5中一致性碱基获得。若max_alleles_consens设置成1(单倍体),则杂合性就修正为0,只对错误率进行估计。其余所有的max_alleles_consens设置都是用于二倍体的(即,两个等位基因)。
以下是step 4中 可能用到的 和必须(带*的)参数:
5. 过滤和获得一致性碱基¶
step4中组装reads得到的估计参数和一个二项式模型估计一致性的等位基因。在该步骤中,过滤了每位点最大允许的Ns(max_Ns_consens)。记录每位点的等位基因数,但过滤的最大碱基数直至step7才会被应用。read深度信息也在此步被保存用于step7中VCF的输出。
以下是step 5中 可能用到的 和必须(带*的)参数:
6. 个体间聚类¶
利用在step3中选择的组装方法对样本件的序列进行聚类。一个等位基因在聚类前随机的进行样本化,以减少歧义字符对聚类的影响,但是结果数据仍保留了部分杂合性信息。聚类后的序列随后用muscle进行组装。
以下是step 6中 可能用到的 和必须(带*的)参数:
7. 过滤和形成结果文件¶
经过过滤器得到最终的组装结果并将其保存为一些列输出文件格式。此步骤常因不同的参数设置而被重复(参数21.min_samples_locus)(见分支部分)
以下是step 6中 可能用到的 和必须(带*的)参数:
- *assembly_name
- *project_dir
- *datatype
- min_samples_locus
- max_Indels_locus
- max_shared_Hs_locus
- max_alleles_consens
- trim_overhang
- output_formats
- pop_assign_file
CLI分支工作流程案例
## 产生参数文件命名为data1;
## 编辑参数文件data1-params.txt
ipyrad -n data1
## 默认参数运行步骤1-2
ipyrad -p params-data1.txt -s 12
## 分支产生‘拷贝’命名为data2
ipyrad -p params-data1.txt -b data2
## 编辑参数文件data2-params.txt,修改相应参数;
## 例如,修改个体内聚类阈值从0.85 至 0.90;
## 运行剩余步骤 (3-7)
ipyrad -p params-data1.txt -s 34567
ipyrad -p params-data2.txt -s 34567
Python API分支工作流程案例
## 输入 ipyrad
import ipyrad as ip
## 产生组装程序并修改参数设置
data1 = ip.Assembly("data1")
data1.set_params("project_dir", "example")
data1.set_params("raw_fastq_path", "data/*.fastq")
data1.set_params("barcodes_path", "barcodes.txt")
## 运行步骤1-2
data1.run("12")
## 产生新的分支并命名为data2;
## 修改参数
data2 = data1.branch("data2")
data2.set_params("clust_threshold", 0.90)
## 运行3-7步
data1.run("34567")
data2.run("34567")